
Inlägg
Inlägg som Tayg har skrivit i forumet
Steered molecular dynamics - problem med att få till "the pulling groups".
(Jag hade ingen aning om vart jag skulle placera denna - ber om ursäkt om den hamnar helt fel!)
På engelska men jag hoppas att det är okej ändå:
I'm trying to perform a steered molecular dynamics simulation where I pull a magnesium ion from two amino acids. The amino acids are on either side of a protein dimer interphase and the magnesium ion is between them. The amino acids both have a COO- group at the end.
I the first run I had the four oxygen atoms as one group and the magnesium ion as the other group. The result was that the magnesium ion was stuck to one of the oxygens through the entire simulation.
In the second run I split the oxygens into four groups and the magnesium ion was the fifth. Now the magnesium is still attached to an oxygen, but the magnesium does leave this site and moves further from it during the simulation.
What I do need it the magnesium ion to be attacted to all the four oxygens and to be pulled away from it.
The starting frame is a pdb-frame where the magnesium ion is positioned almost exactly in the center between all of the oxygens. But the starting frame of the simulation is with the magnesium ion positioned stuck to an oxygen atom.
Any advice on what to adjust?
Regarding spring constant and pulling velocity. I can of course adjust, run and see what happens. But I rather not as I am running on a budget of a computational group where I'm doing an internship. Here it to me doesn't feel like it is the spring constant or pulling velocity that is the issue, as the issue is that the magnesium doesn't seem to be attracted to all oxygens. But could the spring constant and velocity be the issue?
Could the following be the issue and the solution to remove the periodic boundary conditions? From a information page about SMD and non-equilibrium pulling: "In GROMACS, there are three ways to define the center of mass of a group. The standard way is a “plain” center of mass, possibly with additional weighting factors. With periodic boundary conditions it is no longer possible to uniquely define the center of mass of a group of atoms. Therefore, a reference atom is used. For determining the center of mass, for all other atoms in the group, the closest periodic image to the reference atom is used. This uniquely defines the center of mass. By default, the middle (determined by the order in the topology) atom is used as a reference atom, but the user can also select any other atom if it would be closer to center of the group."
I have very little experience in SMD and do not yet understand everything about what I bring up above. I've had bad luck with my supervisor being ill a lot in the past month and later very busy trying to catch up so I haven't had the possibility to ask things e.g. about spring constants or velocity which I don't yet understand. I'm not after someone giving me the answers but hints to what to look at are very much appreciated.
Skrivet av zonar:
Jag har knåpat ganska många rader kod och program för allt ifrån utvärdering av provning, automation av simuleringsflöden till utveckling av utmattningskriterier för härdade stål. Baserat på informationen i din exempeldata behövs lite modifierande av koden för att det ska funka rakt av.
Numpy modulen här läser in filen som om det är text och allt som startar med @ eller# anses vara en kommentar. Det tar med andra ord allt som inte är bortkommenterat och gör om det till en numpy.array (tänk en vektor). Numpy returnerar en vector per kolumn.
Resten av koden är för att skapa figuren samt sätta lite förklaringar på axlarna.
import matplotlib.pyplot as plt
import numpy as np
# Replace to your file names here
force_time_file_path = "force-time.xvg"
displacement_time_file_path = "displacment-time.xvg"
# Load file 1
time, force = np.loadtxt(force_time_file_path ,comments=["@", "#"],unpack=True)
# Load file 2
time, displacement, reference_displacement = np.loadtxt(displacement_time_file_path ,comments=["@", "#"],unpack=True)
# Plot figure
plt.plot(force, displacement, reference_displacement )
plt.xlabel("Force [N]")
plt.ylabel("Displacement [mm]")
plt.savefig("volume.png", format="png", dpi=300)
plt.show()
/z
Stort tack! Och länkarna du skicka i ditt första svar var precis vad jag försökte hitta (men jag villade nog bort mig i vad jag använde för sökord)!
Skrivet av perost:
Du är nog lite ute och cyklar här, import matplotlib.pyplot as plt är ett python-kommando som du skriver i ditt python-script. Det är inget du kan köra direkt i terminalen, utan vad du försöker göra just nu är att installera imagemagick som råkar ha ett program som heter import (ett skärmdumpsprogram).
Men för att lösa problemet du har med apt så behöver du troligtvis göra som kommandot säger och köra apt-get update för att uppdatera dina paketförråd, paketen har troligtvis uppdaterats på servern och stämmer inte längre med vad ditt paketförråd säger. Detta är dock som sagt inget du behöver göra nu, men bara så du vet ifall du faktiskt behöver installera något med apt i framtiden.
Ojdå, tack!
Jag försökte faktiskt med apt-get update men fick då:
apt-get update
Reading package lists... Done
E: Could not open lock file /var/lib/apt/lists/lock - open (13: Permission denied)
E: Unable to lock directory /var/lib/apt/lists/
W: Problem unlinking the file /var/cache/apt/pkgcache.bin - RemoveCaches (13: Permission denied)
W: Problem unlinking the file /var/cache/apt/srcpkgcache.bin - RemoveCaches (13: Permission denied)
Försökte söka på nätet för att hitta något men hittade inget. Vet någon vad detta innebär?
Skrivet av pine-orange:
Du behöver inte installera import, det finns inbyggt i python. Det du behöver göra är att skapa en .py fil och sen köra filen mha python.
Mitt tips är att du kollar på någon video på Youtube som förklarar Pythongrunderna.
Och tack för tipset om Youtube!
Skrivet av pine-orange:
Du behöver inte installera import, det finns inbyggt i python. Det du behöver göra är att skapa en .py fil och sen köra filen mha python.
Så rader som tex:
import matplotlib.pyplot as plt
de ska man alltså inte köra i Ubuntu-rutan?
Är det mha batch man kör ett skript? Så att vad jag ska göra är att göra en textfil med skriptet som jag sparar som .py och sedan köra det med batch <filnamn>.py?
Kan inte installera kommandot import
Försöker installera kommandot import, för att använda för att importera bla numpy. Jag har försökt alla tre alternativen nedan och får felmeddelanden för alla.
Mina frågor är:
Vad är skillnaden mellan de tre alternativen man kan installera import med?
Och hur ska jag tolka felmeddelandet?
(biomolsim) xyz@xyz:/mnt/c/Users/my_username/Desktop/NEWEST_DUMPS/NDXPDB_MAKING_OF_FRAME_49/frame_49_snellius_parametertest_k_1000$ import matplotlib.pyplot as plt
Command 'import' not found, but can be installed with:
sudo apt install graphicsmagick-imagemagick-compat # version 1.4+really1.3.38-1, or
sudo apt install imagemagick-6.q16 # version 8:6.9.11.60+dfsg-1.3build2
sudo apt install imagemagick-6.q16hdri # version 8:6.9.11.60+dfsg-1.3build2
(biomolsim) xyz@xyz:/mnt/c/Users/my_username/Desktop/NEWEST_DUMPS/NDXPDB_MAKING_OF_FRAME_49/frame_49_snellius_parametertest_k_1000$ sudo apt install graphicsmagick-imagemagick-compat
[sudo] password for xyz:
Reading package lists... Done
Building dependency tree... Done
Reading state information... Done
The following additional packages will be installed:
fonts-droid-fallback fonts-noto-mono fonts-urw-base35 ghostscript
graphicsmagick libaom3 libavahi-client3 libavahi-common-data
libavahi-common3 libcups2 libdav1d5 libde265-0 libdeflate0
libgraphicsmagick-q16-3 libgs9 libgs9-common libheif1 libidn12
libijs-0.35 libjbig0 libjbig2dec0 liblcms2-2 libopenjp2-7 libpaper-utils
libpaper1 libtiff5 libwebp7 libwebpmux3 libwmflite-0.2-7 libx265-199
poppler-data
Suggested packages:
fonts-noto fonts-freefont-otf | fonts-freefont-ttf fonts-texgyre
ghostscript-x graphicsmagick-dbg cups-common liblcms2-utils poppler-utils
fonts-japanese-mincho | fonts-ipafont-mincho fonts-japanese-gothic
| fonts-ipafont-gothic fonts-arphic-ukai fonts-arphic-uming fonts-nanum
The following NEW packages will be installed:
fonts-droid-fallback fonts-noto-mono fonts-urw-base35 ghostscript
graphicsmagick graphicsmagick-imagemagick-compat libaom3 libavahi-client3
libavahi-common-data libavahi-common3 libcups2 libdav1d5 libde265-0
libdeflate0 libgraphicsmagick-q16-3 libgs9 libgs9-common libheif1
libidn12 libijs-0.35 libjbig0 libjbig2dec0 liblcms2-2 libopenjp2-7
libpaper-utils libpaper1 libtiff5 libwebp7 libwebpmux3 libwmflite-0.2-7
libx265-199 poppler-data
0 upgraded, 32 newly installed, 0 to remove and 7 not upgraded.
Need to get 183 kB/23.7 MB of archives.
After this operation, 99.8 MB of additional disk space will be used.
Do you want to continue? [Y/n] Y
Ign:1 http://archive.ubuntu.com/ubuntu jammy-updates/main amd64 libtiff5 amd64 4.3.0-6ubuntu0.1
Err:1 http://security.ubuntu.com/ubuntu jammy-updates/main amd64 libtiff5 amd64 4.3.0-6ubuntu0.1
404 Not Found [IP: 91.189.91.38 80]
E: Failed to fetch http://security.ubuntu.com/ubuntu/pool/main/t/tiff/libtiff5_4... 404 Not Found [IP: 91.189.91.38 80]
E: Unable to fetch some archives, maybe run apt-get update or try with --fix-missing?
(biomolsim) xyz@xyz:/mnt/c/Users/my_username/Desktop/NEWEST_DUMPS/NDXPDB_MAKING_OF_FRAME_49/frame_49_snellius_parametertest_k_1000$
Skrivet av pine-orange:
https://gromacswrapper.readthedocs.io/en/latest/installation....
https://stackoverflow.com/a/65725635
Det verkar som att xvg bara är textfiler så du kan ju bara öppna med notepad och se hur de ser ut. Den andra länken borde räcka egentligen.
Tack!!
Skrivet av danneee:
Hittade detta https://www.compchems.com/how-to-extract-and-plot-thermodynam...
Men är inte så haj på python så det kanske inte är exakt vad som efterfrågas..
Tack!!
Skrivet av zonar:
Den såg väldigt lovande ut, detta ser ganska rakt fram ut:
import matplotlib.pyplot as plt
import numpy as np
x,y = np.loadtxt("volume.xvg",comments=["@", "#"],unpack=True)
plt.plot(x,y)
plt.xlabel("time (ps)")
plt.ylabel("volume (nm^3)")
plt.savefig("volume.png", format="png", dpi=300)
plt.show()
Vilket kanske i ditt fall kan modifieras till något liknande
import matplotlib.pyplot as plt
import numpy as np
# Load file 1
f, t = np.loadtxt("force-time.xvg",comments=["@", "#"],unpack=True)
# Load file 2
d, t = np.loadtxt("displacment-time.xvg",comments=["@", "#"],unpack=True)
# Plot figure
plt.plot(f,d)
plt.xlabel("Force [N]")
plt.ylabel("Displacement [mm]")
plt.savefig("volume.png", format="png", dpi=300)
plt.show()
Ingen av modulerna numpy och matplotlib följer med i installationen av Python utan dessa behöver först installeras. Kolla på respektive sida för instruktioner om det inte redan är fixat.
/z
Sort tack för alla svar! Ska kolla igenom och se om jag kan förstå det och få till detta
Hur gör du, förstår du allt du skriver och varför du skriver det, eller använder många mycket färdiga bitar (tex genom sökande i forum) som man pusslar ihop?
Börjar den automatiskt att läsa av där # och @ slutar (jag ser en bit som jag eventuellt kan tolka som att # och @ ska tolkas som kommentarer) eller bör jag kopiera över själva x och y raderna i något nytt dokument?
Nedan är översta delen av de två filerna. Tiden fortsätter upp till 10 000 ns.
# This file was created Sat Nov 26 22:15:38 2022
# Created by:
# GROMACS - gmx mdrun, 2021.6-EasyBuild-4.6.2 (-:
#
# Executable: /sw/arch/RHEL8/EB_production/2022/software/GROMACS/2021.6-foss-2022a/bin/gmx_mpi
# Data prefix: /sw/arch/RHEL8/EB_production/2022/software/GROMACS/2021.6-foss-2022a
# Working dir: /gpfs/scratch1/nodespecific/tcn370/evanleuven.1875753
# Command line:
# gmx_mpi mdrun -deffnm md -v -ntomp 1 -maxh 3
# gmx mdrun is part of G R O M A C S:
#
# GROningen MAchine for Chemical Simulation
#
@ title "Pull force"
@ xaxis label "Time (ps)"
@ yaxis label "Force (kJ/mol/nm)"
@TYPE xy
0.0000 70.0374
0.1000 75.4116
0.2000 81.2423
0.3000 33.8946
0.4000 4.1915
0.5000 16.3492
0.6000 16.4572
0.7000 17.499
0.8000 31.6646
0.9000 41.5101
1.0000 53.8529
1.1000 59.7578
1.2000 61.4971
1.3000 74.6553
1.4000 58.7796
1.5000 53.4414
1.6000 23.5608
1.7000 14.921
1.8000 14.9907
1.9000 -4.31798
2.0000 0.875456
2.1000 12.201
2.2000 29.2113
2.3000 12.5113
2.4000 28.7196
2.5000 20.3133
2.6000 3.54749
2.7000 -3.36847
2.8000 -2.53457
2.9000 22.9303
3.0000 23.4295
3.1000 3.36555
3.2000 9.77809
3.3000 5.79202
3.4000 -0.810893
3.5000 -17.9361
# This file was created Sat Nov 26 22:15:38 2022
# Created by:
# GROMACS - gmx mdrun, 2021.6-EasyBuild-4.6.2 (-:
#
# Executable: /sw/arch/RHEL8/EB_production/2022/software/GROMACS/2021.6-foss-2022a/bin/gmx_mpi
# Data prefix: /sw/arch/RHEL8/EB_production/2022/software/GROMACS/2021.6-foss-2022a
# Working dir: /gpfs/scratch1/nodespecific/tcn370/evanleuven.1875753
# Command line:
# gmx_mpi mdrun -deffnm md -v -ntomp 1 -maxh 3
# gmx mdrun is part of G R O M A C S:
#
# GROningen MAchine for Chemical Simulation
#
@ title "Pull COM"
@ xaxis label "Time (ps)"
@ yaxis label "Position (nm)"
@TYPE xy
@ view 0.15, 0.15, 0.75, 0.85
@ legend on
@ legend box on
@ legend loctype view
@ legend 0.78, 0.8
@ legend length 2
@ s0 legend "1"
@ s1 legend "1 ref"
0.0000 3.92996 4
0.1000 3.9246 4.00002
0.2000 3.91879 4.00003
0.3000 3.96615 4.00005
0.4000 3.99587 4.00006
0.5000 3.98373 4.00008
0.6000 3.98363 4.00009
0.7000 3.98261 4.00011
0.8000 3.96846 4.00012
0.9000 3.95862 4.00014
1.0000 3.9463 4.00015
1.1000 3.94041 4.00017
1.2000 3.93868 4.00018
1.3000 3.92554 4.0002
1.4000 3.94143 4.00021
1.5000 3.94678 4.00023
1.6000 3.97668 4.00024
1.7000 3.98533 4.00026
1.8000 3.98528 4.00027
1.9000 4.0046 4.00029
2.0000 3.99942 4.0003
2.1000 3.98811 4.00032
2.2000 3.97112 4.00033
2.3000 3.98783 4.00035
2.4000 3.97164 4.00036
2.5000 3.98006 4.00038
2.6000 3.99684 4.00039
2.7000 4.00377 4.00041
2.8000 4.00295 4.00042
2.9000 3.9775 4.00044
3.0000 3.97702 4.00045
3.1000 3.9971 4.00047
3.2000 3.9907 4.00048
3.3000 3.9947 4.0005
3.4000 4.00132 4.00051
3.5000 4.01846 4.00053
Python, Numpy, .xvg-filer - göra graf mha två .xvg filer
(Ber om ursäkt om detta inte var korrekt Forum!)
Jag gör ett "internship" med steered molecular dynamics. I kort ett det är kasst upplägg med en handledare som är delvis pappaledig och som mest kommunicerar via en chatfunktion, där han knappt svarar alls.
Min uppgift är att göra en graf. Jag ska använda:
Python och Numpy
tillsammans med:
Två .xvg filer, där den ena har kraft (force) vs tid och den andra avstånd mot tid. Slutgrafen ska ha kraft mot avstånd.
Handledarens ord förutom det ovan är "googla runt, stackoverflow.com har mycket".
Han vet mycket väl att jag inte har någon erfarenhet av Python eller Numpy. Jag har inte hittat det jag ska göra i någon tråd och jag vet inte vart jag ska börja om jag ska göra detta själv.
Tips om bra sida att kolla på, hur jag ska gå tillväga för att försöka komma någonstans, eller annat mottages tacksamt!
GROMACS: gmx pdb2gmx, fatal error.
Posten är på engelska (har skrivit samma i ett annat forum, på engelska) men svar på svenska/engelska funkar lika bra.
GROMACS version: 2021.4-Ubuntu-2021.4-2
GROMACS modification: Yes/No (don’t know what this means)
I am new to this so I don’t know what information you need to at all be able to try and answer my question. Please ask if something is missing.
I’ve been visualising an enzyme in VMD from a gro-file. The enzyme has the ions MG, NA and ZN around it.
It had 4 chains from the beginning and ions I didn’t want to keep. So I used:
gmx make_ndx -f .gro and got an index file.
I made a group with what I wanted to keep and in the following command I used this to make a new gro-file:
gmx trjconv -s md_nowater.tpr -f .gro -n index.ndx -o .pdb
Then I took this pdb file to go:
gmx pdb2gmx -f .pdb -o .gro -water tip3p
I come to the step where you should chose the force field and chose amber99sb-ildn. And I get the following, fatal, error:
Program: gmx pdb2gmx, version 2021.4-Ubuntu-2021.4-2
Source file: src/gromacs/gmxpreprocess/pdb2gmx.cpp (line 790)
Fatal error:
Atom HE2 in residue HIS 355 was not found in rtp entry HID with 17 atoms
while sorting atoms.
For a hydrogen, this can be a different protonation state, or it
might have had a different number in the PDB file and was rebuilt
(it might for instance have been H3, and we only expected H1 & H2).
Note that hydrogens might have been added to the entry for the N-terminus.
Remove this hydrogen or choose a different protonation state to solve it.
Option -ignh will ignore all hydrogens in the input.
For more information and tips for troubleshooting, please check the GROMACS
website at Common Errors — GROMACS webpage https://www.gromacs.org documentation
I don’t know what this means or how to solve it.
I am following instructions from a supervisor but having said that, not everything has been correct so far as he hasn’t previously done these steps in his research himself.
Skrivet av Yoshman:
OBS: kör allt nedan i en helt ren Ubuntu 22.04 installation i Docker, därför jag kör som "root"...
Där det står "/root/" kommer det på en "riktig" installation vara /home/<username>/
Från nedladdning. OBS: kör på en Mac så använder ARM64, i ditt fall ska det nog vara denna som ska laddas ned
https://repo.anaconda.com/archive/Anaconda3-2022.10-Linux-x86...
root@dcc0d3fbbc11:~# wget https://repo.anaconda.com/archive/Anaconda3-2022.10-Linux-aar...
root@dcc0d3fbbc11:~# sh Anaconda3-2022.10-Linux-aarch64.sh
Edit: @Tayg detta steg går fel i ditt fall om man tittar på första post. När du kör "wget" hamnar ju filen i katalogen du står vilket i ditt fall är hemkatalogen och inte ~/Downloads!!
Man får ju en del frågor under installationen av Conda, vad svarade du t.ex. på denna?
Do you wish the installer to initialize Anaconda3
by running conda init? [yes|no]
[no] >>>
Svarar man "yes" får man detta
modified /root/anaconda3/condabin/conda
modified /root/anaconda3/bin/conda
modified /root/anaconda3/bin/conda-env
no change /root/anaconda3/bin/activate
no change /root/anaconda3/bin/deactivate
no change /root/anaconda3/etc/profile.d/conda.sh
no change /root/anaconda3/etc/fish/conf.d/conda.fish
no change /root/anaconda3/shell/condabin/Conda.psm1
no change /root/anaconda3/shell/condabin/conda-hook.ps1
no change /root/anaconda3/lib/python3.9/site-packages/xontrib/conda.xsh
no change /root/anaconda3/etc/profile.d/conda.csh
modified /root/.bashrc
==> For changes to take effect, close and re-open your current shell. <==
If you'd prefer that conda's base environment not be activated on startup,
set the auto_activate_base parameter to false:
conda config --set auto_activate_base false
Thank you for installing Anaconda3!
===========================================================================
Working with Python and Jupyter is a breeze in DataSpell. It is an IDE
designed for exploratory data analysis and ML. Get better data insights
with DataSpell.
DataSpell for Anaconda is available at: https://www.anaconda.com/dataspell
Antar att du loggat ut och sedan loggat in igen (eller manuellt kört ~/.bashrc)?
När Conda är aktiverat ska du se detta (jag aktiverade det genom att manuellt köra ~/.bashrc)
root@dcc0d3fbbc11:~# which conda
root@dcc0d3fbbc11:~# . ~/.bashrc
(base) root@dcc0d3fbbc11:~#
Notera "(base) ..." prefixet, det säger att jag nu initierat Conda och befinner mig i "base" miljön
I det läget hittas "conda" binären och du kan köra vidare
(base) root@dcc0d3fbbc11:~# which conda
/root/anaconda3/bin/conda
När man kör python på detta sätt använder man inte heller den python som eventuellt redan finns installerad, man använder den som kommer med Anaconda
(base) root@dcc0d3fbbc11:~# which python
/root/anaconda3/bin/python
Stort tack för att du tog dig tid att skriva ihop detta!! Jag valde till slut nu (skulle in och skriva att problemet var löst) att avinstallera det jag installerat enligt lärarens instruktioner och följa de instruktioner som finns på nedladdningssidan. Nu funkar allt som det ska.
Skrivet av perost:
Du laddar ner installationsfilen till din home-mapp (d.v.s. ~) men försöker köra den från ~/Downloads. Ta bort Downloads från sökvägen så borde det gå bättre.
Att which python inte gav något beror på att den i Ubuntu heter python3 istället. Du kan installera python-is-python3 (sudo apt install python-is-python3) om du vill ha ett alias så att du bara kan anropa python istället.
Det finns för övrigt officiella instruktioner för att installera Anaconda.
Dock förstår jag inte den lösningen på "conda command not found" (inte riktat till dig, utan till länken med hjälp till att lösa felmeddelandet). Jag har ju redan Anaconda3. Jag testade ditt tips men kommer till att det redan är installerat. Jag testade att lägga till "-u" och uppdaterade, vilket inte hjälpte heller med "conda" problemet.
which python:
TACK!!
Jo jag vet. Men jag följde lärarens instruktioner för jag tänkte att det väl måste finnas någon poäng med att hon inte bara hänvisade till länken. Men skulle kanske inte gjort det... Hon verkar inte riktigt ha koll på allt i sin instruktion...
Skrivet av Emma242:
Jag har haft sjukt mycket problem med de senaste versionerna av Anaconda i Linux. Senaste året har typ ingenting fungerat, och det blir allt sämre i Windows också.
Det finns säkert lösningar på dina problem, men jag varnar för att det kan vara ett rejält kaninhål. Bioconda går t.ex knappt att köra på nya Linux-distar pga kasst underhåll.
Jag arbetar bland annat med bioinformatiska analyser, vilket jag förstår att du ska försöka dig på? I så fall rekommenderar jag att försöka hitta färdiga Docker-bilder i stället, alternativt skippa virtuella miljöer och installera programmen separat i stället. I de flesta fall behöver du typ bara R och Python. Jag och många av mina kollegor har alltmer övergett Anaconda.
Tack för tipsen! Tyvärr måste jag använda Linux, Ubuntu, Anaconda3 och Python (jag är inte hemma i detta så vet knappt vad jag räknar upp men de programmen ska användas enligt instruktionerna, och läraren). Det är "biomolecular simulations" jag ska göra mha VMD, snellius (supercomputer), och bla dessa program.
Ska dock helt klart ta med dina tips till läraren och speciellt den PhD jag jobbar med
Skrivet av pacc:
Anaconda/conda sätter upp en egen miljö med python och libraries färdiginstallerat.
Att bara installera conda gör ingenting men när du har det bör du kunna köra kommandot och köra kursens pythonmmiljö i en terminal.
Jag förstod inte ditt svar. Men jag måste installera Anaconda3 och använda Ubuntu i kursen. Anaconda3 verkar vara installerat men conda kommandot funkar inte.
conda command not found
Jag försökte följa länken nedan för att lösa felmeddelandet. Kom genom första delen men sedan så får jag igen ett felmeddelande.
Instruktioner:
https://linuxhint.com/conda-command-not-found/
Resultatet:

Så min fråga är om någon vet hur jag kommer vidare.
Varför jag hamnade här var då jag försökte följa instruktioner i kursen där man ska göra det som står på bilden nedan. När jag skrev in det röda (efter att ha gjort resten ovan) så fick jag "conda command not found".
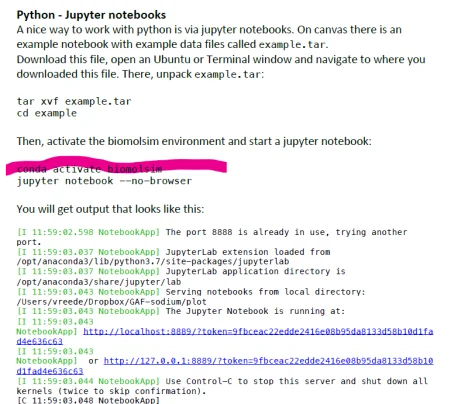
Det som jag innan dess hade gjort var det nedan. Men "which python" gav inget, inte heller ett felmeddelande. "ls" gav Anaconda3. Efter att ha följt länken ovan (linuxhint...) så ger "ls" : Anaconda3-2022.05-Linux-x86_64.sh Anaconda3-2022.05-Linux-x86_64.sh.1 anaconda3.

Nej, inte när jag installerade och inte när jag öppnade det nya fönstret (då fick jag bara koden under EDIT). När den installerade, innan jag stängde fönstret för att öppna det nya så gick allt utan problem. Jag kom fram till xxx@yyy:~$ igen, stängde då fönstret och öppnade det nya.
Anaconda och Python i Ubuntu
(Hoppas jag satte tråden rätt, ber om ursäkt annars!)
Fotnot: jag är verkligen inte hemma i detta, så inga krångliga ord and keep it basic.
Instruktionerna till denna delen:

Kurs på Universitet i simuleringar med biomolekyler.
Jag har installerat Ubuntu, laddat ner anaconda, installerat, stängt terminal fönstret, öppnat ett nytt, skrivit "which python", men jag får inte "the output" ovanför det blå strecken i bilden ovan. Jag får inte heller ett felmeddelande utan det här inget när jag trycker på enter.
Help?
EDIT:
Detta är vad jag får i det nya fönstret.
taygetos@Lenovo88:~$ which python
taygetos@Lenovo88:~$ cd
taygetos@Lenovo88:~$ ls
anaconda3
taygetos@Lenovo88:~$ cd /mnt
taygetos@Lenovo88:/mnt$ ls
c wsl
taygetos@Lenovo88:/mnt$
Skrivet av Lodisen:
En 5800U drar ~15W, medans en 5600H drar upp mot 50W eller så. När den matas med mer kräm höjs hastigheten.
Ja, throttling är att den klockas ned av att den går för varm.
CPUn är inte "sämre". Det är tänkt för olika segment.
5600H har sex kärnor men drar 3x så mycket ström, så dessa sex kärnor körs på högre hastighet än de åtta kärnorna som finns i 5800U. Siffrorna i modellen kan vara missvisande. H-modeller är alltså törstigare än U, som är gjord för att dra lite ström, ge bättre batteritid och kunna sitta i ultraportabla laptops. De kan helt enkelt inte trycka in en H-modell i slimdatorn, då det inte finns tillräckligt med kylning.
Yoga slimmen är så tunn så att den har sämre kylning. CPUn går upp mot 90C i Yoga slimmen, medans den bara når ~66c i Ideapadden. Då kan den hålla högre hastigheter längre.
Om du inte vill kompromissa kan du skaffa en Ideapad 5 med en 5800H-cpu.
Stort tack!!
Hårdvara
Mjukvara
Övrigt
- Igår Tävling: Dela semesterfoton och belönas med presentkort! 27
- Igår SweClockers Noobbyggarguide - Del 5: Kabelkrångel 2
- 3 / 6 Microsoft miljardsatsar på svensk sjukvård 57
- 3 / 6 Året var 2003 – Innan SSD:n vandrade på jorden fanns Raptorn 72
- 3 / 6 Blocket inför BankID-verifiering för alla användare 41
Datorkomponenter
Ljud, bild och kommunikation
- Finns det gaming laptops som inte låter som flygplan?3
- Intel visar Lunar Lake – "den mest effektiva arkitekturen någonsin"96
- Gammal Windows 7-dator börjar bete sig konstigt13
- Apple uppgav fel antal kärnor för Ipad Airs gpu6
- Hjälp med att installera windows 1119
- Krönika: När datorn endast blivit ett jobbverktyg37
- WD Black 770 gen 4 NVME går varm, dålig kylning eller defekt disk?1
- Tråden för 2-kanals stereo med högtalare667
- PizzaClockers - pizzans hemligheter!1442
- Vilket VR-headset ska jag köpa?904
- Säljes Gaming dator
- Säljes Gaming Dator plus skärm
- Säljes Dual 10gbit! HPE ethernet 10gb 2-port 530t adapter. 2st kort.
- Säljes DDR3 + SSD 850 EVO M.2
- Säljes MacBook M1 Pro med tillbehör
- Säljes iPhone 12 Pro Max (256 GB, Pacific Blue)
- Köpes Amd 6800 nvidia 3080 eller bättre köpes
- Säljes Grafikkort 1060, Chassi och PSU 650w
- Säljes SteelSeries Arctis Nova Pro, utan gamedac
- Säljes ROG MAXIMUS Z690 HERO GSKILL RAM
Nytt i testlabbet
Tester av chassi, grafikkort, processorer m.m.
- SweClockers Noobbyggarguide - Del 5: Kabelkrångel2
- SweClockers Noobbyggarguide - Del 4: Aggressivt aggregat8
- Årets bästa soundbar – vi väljer favoriterna78
- Snabba upp din gamla dator med uppgradering66
- SweClockers Noobbyggarguide - Del 3: Den dramatiska moderkortsmonteringen12
- Snabbtest: Steam Deck överraskar som arbetsdator31
- Snabbtest: 3DMark Steel Nomad – visuellt avancerat, förvånansvärt lättdrivet80
- Grafikprestanda i Senua's Saga: Hellblade II – ett av de tyngsta PC-spelen någonsin187
- Grafikprestanda i Ghost of Tsushima – en välpolerad PC-version28
- SweClockers Noobbyggarguide Del 213
